
Contains Nonbinding Recommendations
Draft – Not for Implementation
Computer Software Assurance for
Production and Quality System
Software
______________________________________________________________________________
Draft Guidance for Industry and
Food and Drug Administration Staff
DRAFT GUIDANCE
This draft guidance document is being distributed for comment purposes
only.
Document issued on September 13, 2022.
You should submit comments and suggestions regarding this draft document within 60 days of
publication in the Federal Register of the notice announcing the availability of the draft
guidance. Submit electronic comments to https://www.regulations.gov. Submit written
comments to the Dockets Management Staff, Food and Drug Administration, 5630 Fishers Lane,
Room 1061, (HFA-305), Rockville, MD 20852. Identify all comments with the docket number
listed in the notice of availability that publishes in the Federal Register.
For questions about this document regarding CDRH-regulated devices, contact the Compliance
and Quality Staff at 301-796-5577 or by email at [email protected]. For questions
about this document regarding CBER-regulated devices, contact the Office of Communication,
Outreach, and Development (OCOD) at 1-800-835-4709 or 240-402-8010, or by email at
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Devices and Radiological Health
Center for Biologics Evaluation and Research

Contains Nonbinding Recommendations
Draft – Not for Implementation
Preface
Additional Copies
CDRH
Additional copies are available from the Internet. You may also send an email request to CDRH-
[email protected] to receive a copy of the guidance. Please include the document number
17045 and complete title of the guidance in the request.
CBER
Additional copies are available from the Center for Biologics Evaluation and Research (CBER),
Office of Communication, Outreach, and Development (OCOD), 10903 New Hampshire Ave.,
Bldg. 71, Room 3128, Silver Spring, MD 20993-0002, or by calling 1-800-835-4709 or 240-402-
biologics/guidance-compliance-regulatory-information-biologics/biologics-guidances.

Contains Nonbinding Recommendations
Draft – Not for Implementation
Table of Contents
I. Introduction .......................................................................................................................................................... 4
II. Background .......................................................................................................................................................... 5
III. Scope .................................................................................................................................................................... 6
IV. Computer Software Assurance ............................................................................................................................. 6
V. Computer Software Assurance Risk Framework ................................................................................................. 7
Identifying the Intended Use........................................................................................................................7
Determining the Risk-Based Approach........................................................................................................9
Determining the Appropriate Assurance Activities ...................................................................................13
Establishing the Appropriate Record .........................................................................................................16
Appendix A. Examples ................................................................................................................................................ 20
Example 1: Nonconformance Management System ............................................................................................... 20
Example 2: Learning Management System (LMS) ................................................................................................ 23
Example 3: Business Intelligence Applications ...................................................................................................... 24
Contains Nonbinding Recommendations
Draft – Not for Implementation
4
Computer Software Assurance for
1
Production and Quality System
2
Software
3
______________________________________________________________________________
4
Draft Guidance for Industry and
5
Food and Drug Administration Staff
6
7
This draft guidance, when finalized, will represent the current thinking of the Food and Drug
8
Administration (FDA or Agency) on this topic. It does not establish any rights for any person
9
and is not binding on FDA or the public. You can use an alternative approach if it satisfies
10
the requirements of the applicable statutes and regulations. To discuss an alternative
11
approach, contact the FDA staff or Office responsible for this guidance as listed on the title
12
page.
13
14
I. Introduction
1
15
FDA is issuing this draft guidance to provide recommendations on computer software assurance
16
for computers and automated data processing systems used as part of medical device production
17
or the quality system. This draft guidance is intended to:
18
19
· Describe “computer software assurance” as a risk-based approach to establish confidence
20
in the automation used for production or quality systems, and identify where additional
21
rigor may be appropriate; and
22
23
· Describe various methods and testing activities that may be applied to establish computer
24
software assurance and provide objective evidence to fulfill regulatory requirements,
25
such as computer software validation requirements in 21 CFR part 820 (Part 820).
26
27
When final, this guidance will supplement FDA’s guidance, “General Principles of Software
28
Validation” (“Software Validation guidance”)
2
except this guidance will supersede Section 6
29
(“Validation of Automated Process Equipment and Quality System Software”) of the Software
30
Validation guidance.
31
32
1
This guidance has been prepared by the Center for Devices and Radiological Health (CDRH) and the Center for
Biologics Evaluation and Research (CBER) in consultation with the Center for Drug Evaluation and Research
(CDER), Office of Combination Products (OCP), and Office of Regulatory Affairs (ORA).
2
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principles-
software-validation.

Contains Nonbinding Recommendations
Draft – Not for Implementation
5
For the current edition of the FDA-recognized consensus standard referenced in this document,
33
see the FDA Recognized Consensus Standards Database.
3
34
35
In general, FDA’s guidance documents do not establish legally enforceable responsibilities.
36
Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only
37
as recommendations, unless specific regulatory or statutory requirements are cited. The use of
38
the word should in Agency guidances means that something is suggested or recommended, but
39
not required.
40
41
II. Background
42
FDA envisions a future state where the medical device ecosystem is inherently focused on device
43
features and manufacturing practices that promote product quality and patient safety. FDA has
44
sought to identify and promote successful manufacturing practices and help device
45
manufacturers raise their manufacturing quality level. In doing so, one goal is to help
46
manufacturers produce high-quality medical devices that align with the laws and regulations
47
implemented by FDA. Compliance with the Quality System regulation, Part 820, is required for
48
manufacturers of finished medical devices to the extent they engage in operations to which Part
49
820 applies. The Quality System regulation includes requirements for medical device
50
manufacturers to develop, conduct, control, and monitor production processes to ensure that a
51
device conforms to its specifications (21 CFR 820.70, Production and Process Controls),
52
including requirements for manufacturers to validate computer software used as part of
53
production or the quality system for its intended use (see 21 CFR 820.70(i)).
4
Recommending
54
best practices should promote product quality and patient safety, and correlate to higher-quality
55
outcomes. This draft guidance addresses practices relating to computers and automated data
56
processing systems used as part of production or the quality system.
57
58
In recent years, advances in manufacturing technologies, including the adoption of automation,
59
robotics, simulation, and other digital capabilities, have allowed manufacturers to reduce sources
60
of error, optimize resources, and reduce patient risk. FDA recognizes the potential for these
61
technologies to provide significant benefits for enhancing the quality, availability, and safety of
62
medical devices, and has undertaken several efforts to help foster the adoption and use of such
63
technologies.
64
65
Specifically, FDA has engaged with stakeholders via the Medical Device Innovation Consortium
66
(MDIC), site visits to medical device manufacturers, and benchmarking efforts with other
67
industries (e.g., automotive, consumer electronics) to keep abreast of the latest technologies and
68
to better understand stakeholders’ challenges and opportunities for further advancement. As part
69
of these ongoing efforts, medical device manufacturers have expressed a desire for greater clarity
70
regarding the Agency’s expectations for software validation for computers and automated data
71
processing systems used as part of production or the quality system. Given the rapidly changing
72
3
Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm.
4
This guidance discusses the “intended use” of computer software used as part of production or the quality system
(see 21 CFR 820.70(i)), which is different from the intended use of the device itself (see 21 CFR 801.4).

Contains Nonbinding Recommendations
Draft – Not for Implementation
6
nature of software, manufacturers have also expressed a desire for a more iterative, agile
73
approach for validation of computer software used as part of production or the quality system.
74
75
Traditionally, software validation has often been accomplished via software testing and other
76
verification activities conducted at each stage of the software development lifecycle. However,
77
as explained in FDA’s Software Validation guidance, software testing alone is often insufficient
78
to establish confidence that the software is fit for its intended use. Instead, the Software
79
Validation guidance recommends that “software quality assurance” focus on preventing the
80
introduction of defects into the software development process, and it encourages use of a risk-
81
based approach for establishing confidence that software is fit for its intended use.
82
83
FDA believes that applying a risk-based approach to computer software used as part of
84
production or the quality system would better focus manufacturers’ assurance activities to help
85
ensure product quality while helping to fulfill the validation requirements of 21 CFR 820.70(i).
86
For these reasons, FDA is now providing recommendations on computer software assurance for
87
computers and automated data processing systems used as part of medical device production or
88
the quality system. FDA believes that these recommendations will help foster the adoption and
89
use of innovative technologies that promote patient access to high-quality medical devices and
90
help manufacturers to keep pace with the dynamic, rapidly changing technology landscape, while
91
promoting compliance with laws and regulations implemented by FDA.
92
93
III. Scope
94
When final, this guidance is intended to provide recommendations regarding computer software
95
assurance for computers or automated data processing systems used as part of production or the
96
quality system.
97
98
This guidance is not intended to provide a complete description of all software validation
99
principles. FDA has previously outlined principles for software validation, including managing
100
changes as part of the software lifecycle, in FDA’s Software Validation guidance.
This guidance
101
applies the risk-based approach to software validation discussed in the Software Validation
102
guidance to production or quality system software. This guidance additionally discusses specific
103
risk considerations, acceptable testing methods, and efficient generation of objective evidence
104
for production or quality system software.
105
106
This guidance does not provide recommendations for the design verification or validation
107
requirements specified in 21 CFR 820.30 when applied to software in a medical device (SiMD)
108
or software as a medical device (SaMD). For more information regarding FDA’s
109
recommendations for design verification or validation of SiMD or SaMD, see the Software
110
Validation guidance.
111
112
IV. ComputerSoftwareAssurance
113
Computer software assurance is a risk-based approach for establishing and maintaining
114
confidence that software is fit for its intended use. This approach considers the risk of
115

Contains Nonbinding Recommendations
Draft – Not for Implementation
7
compromised safety and/or quality of the device (should the software fail to perform as intended)
116
to determine the level of assurance effort and activities appropriate to establish confidence in the
117
software. Because the computer software assurance effort is risk-based, it follows a least-
118
burdensome approach, where the burden of validation is no more than necessary to address the
119
risk. Such an approach supports the efficient use of resources, in turn promoting product quality.
120
121
In addition, computer software assurance establishes and maintains that the software used in
122
production or the quality system is in a state of control throughout its lifecycle (“validated
123
state”). This is important because manufacturers increasingly rely on computers and automated
124
processing systems to monitor and operate production, alert responsible personnel, and transfer
125
and analyze production data, among other uses. By allowing manufacturers to leverage
126
principles such as risk-based testing, unscripted testing, continuous performance monitoring, and
127
data monitoring, as well as validation activities performed by other entities (e.g., developers,
128
suppliers), the computer software assurance approach provides flexibility and agility in helping
129
to assure that the software maintains a validated state consistent with 21 CFR 820.70(i).
130
131
Software that is fit for its intended use and that maintains a validated state should perform as
132
intended, helping to ensure that finished devices will be safe and effective and in compliance
133
with regulatory requirements (see 21 CFR 820.1(a)(1)). Section V below outlines a risk-based
134
framework for computer software assurance.
135
136
V. ComputerSoftwareAssuranceRiskFramework
137
The following approach is intended to help manufacturers establish a risk-based framework for
138
computer software assurance throughout the software’s lifecycle. Examples of applying this risk
139
framework to various computer software assurance situations are provided in Appendix A.
140
IdentifyingtheIntendedUse
141
The regulation requires manufacturers to validate software that is used as part of production or
142
the quality system for its intended use (see 21 CFR 820.70(i)). To determine whether the
143
requirement for validation applies, manufacturers must first determine whether the software is
144
intended for use as part of production or the quality system.
145
146
In general, software used as part of production or the quality system falls into one of two
147
categories: software that is used directly as part of production or the quality system, and software
148
that supports production or the quality system.
149
150
Software with the following intended uses are considered to be used directly as part of
151
production or the quality system:
152
153
· Software intended for automating production processes, inspection, testing, or the
154
collection and processing of production data; and
155
· Software intended for automating quality system processes, collection and processing of
156
quality system data, or maintaining a quality record established under the Quality System
157
regulation.
158

Contains Nonbinding Recommendations
Draft – Not for Implementation
8
159
Software with the following intended uses are considered to be used to support production or
160
the quality system:
161
162
· Software intended for use as development tools that test or monitor software systems or
163
that automate testing activities for the software used as part of production or the quality
164
system, such as those used for developing and running scripts; and
165
· Software intended for automating general record-keeping that is not part of the quality
166
record.
167
168
Both kinds of software are used as “part of” production or the quality system and must be
169
validated under 21 CFR 820.70(i). However, as further discussed below, supporting software
170
often carries lower risk, such that under a risk-based computer software assurance approach, the
171
effort of validation may be reduced accordingly without compromising safety.
172
173
On the other hand, software with the following intended uses generally are not considered to be
174
used as part of production or the quality system, such that the requirement for validation in 21
175
CFR 820.70(i) would not apply:
176
177
· Software intended for management of general business processes or operations, such as
178
email or accounting applications; and
179
· Software intended for establishing or supporting infrastructure not specific to production
180
or the quality system, such as networking or continuity of operations.
181
182
FDA recognizes that software used in production or the quality system is often complex and
183
comprised of several features, functions, and operations;
5
software may have one or more
184
intended uses depending on the individual features, functions, and operations of that software. In
185
cases where the individual features, functions, and operations have different roles within
186
production or the quality system, they may present different risks with different levels of
187
validation effort. FDA recommends that manufacturers examine the intended uses of the
188
individual features, functions, and operations to facilitate development of a risk-based assurance
189
strategy. Manufacturers may decide to conduct different assurance activities for individual
190
features, functions, or operations.
191
192
For example, a commercial off-the-shelf (COTS) spreadsheet software may be comprised of
193
various functions with different intended uses. When utilizing the basic input functions of the
194
COTS spreadsheet software for an intended use of documenting the time and temperature
195
readings for a curing process, a manufacturer may not need to perform additional assurance
196
activities beyond those conducted by the COTS software developer and initial installation and
197
configuration. The intended use of the software, “documenting readings,” only supports
198
maintaining the quality system record and poses a low process risk. As such, initial activities
199
5
That is, software is often an integration of “features,” that are used together to perform a “function” that provides a
desired outcome. Several functions of the software may, in turn, be applied together in an “operation” to perform
practical work in a process. For the purposes of this guidance, a “function” refers to a “software function” and is not
to be confused with a “device function.”

Contains Nonbinding Recommendations
Draft – Not for Implementation
9
such as the vendor assessment and software installation and configuration may be sufficient to
200
establish that the software is fit for its intended use and maintains a validated state. However, if a
201
manufacturer utilizes built-in functions of the COTS spreadsheet to create custom formulas that
202
are directly used in production or the quality system, then additional risks may be present. For
203
example, if a custom formula automatically calculates time and temperature statistics to monitor
204
the performance and suitability of the curing process, then additional validation by the
205
manufacturer might be necessary.
206
207
For the purposes of this guidance, we describe and recommend a computer software assurance
208
framework by examining the intended uses of the individual features, functions, or operations of
209
the software. However, in simple cases where software only has one intended use (e.g., if all of
210
the features, functions, and operations within the software share the same intended use),
211
manufacturers may not find it helpful to examine each feature, function, and operation
212
individually. In such cases, manufacturers may develop a risk-based approach and consider
213
assurance activities based on the intended use of the software overall.
214
215
FDA recommends that manufacturers document their decision-making process for determining
216
whether a software feature, function, or operation is intended for use as part of production or the
217
quality system in their Standard Operating Procedures (SOPs).
218
219
DeterminingtheRiskBasedApproach
220
Once a manufacturer has determined that a software feature, function, or operation is intended
221
for use as part of production or the quality system, FDA recommends using a risk-based analysis
222
to determine appropriate assurance activities. Broadly, this risk-based approach entails
223
systematically identifying reasonably foreseeable software failures, determining whether such a
224
failure poses a high process risk, and systematically selecting and performing assurance activities
225
commensurate with the medical device or process risk, as applicable.
226
227
Note that conducting a risk-based analysis for computer software assurance for production or
228
quality system software is distinct from performing a risk analysis for a medical device as
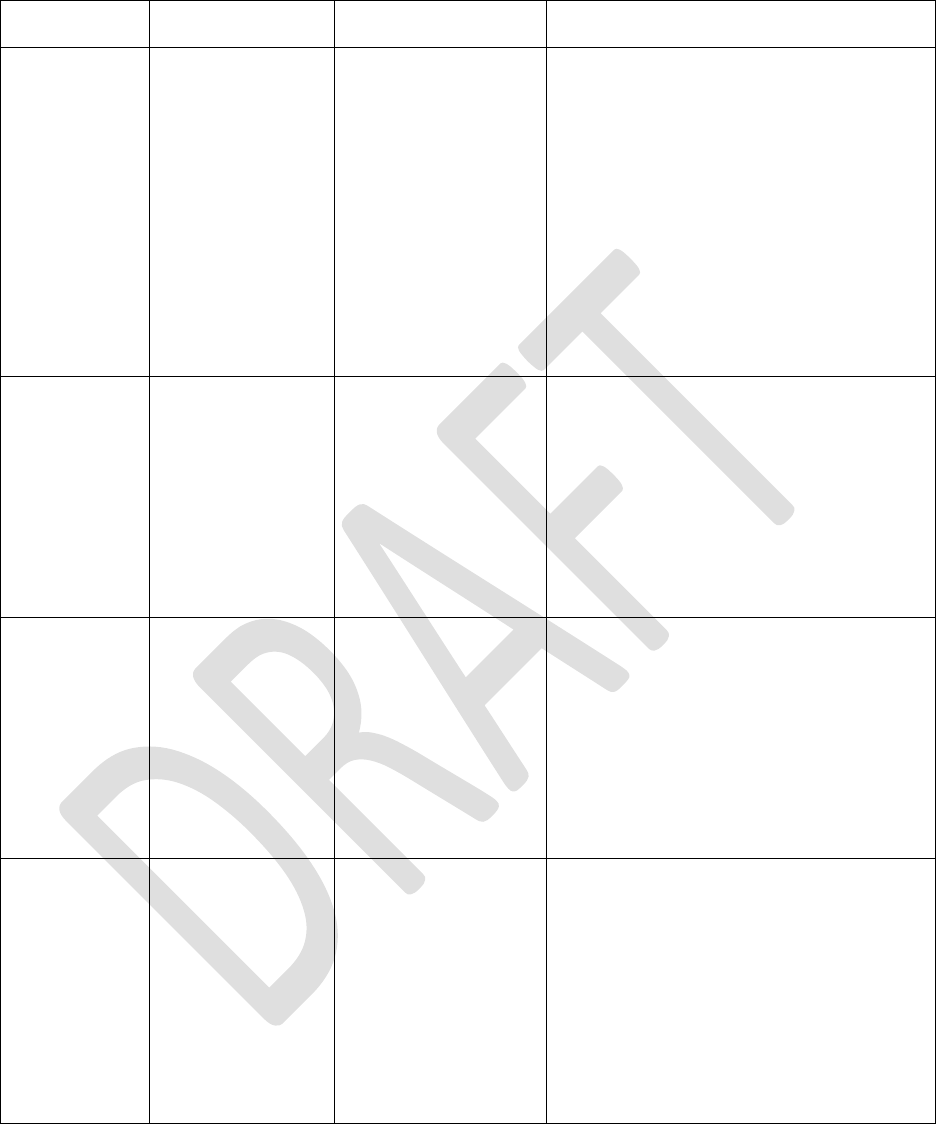
229
described in ISO 14971:2019 – Medical devices – Application of risk management to medical
230
devices. Unlike the risks contemplated in ISO 14971:2019 for analysis (medical device risks),
231
failures of the production or the quality system software to perform as intended do not occur in a
232
probabilistic manner where an assessment for the likelihood of occurrence for a particular risk
233
could be estimated based on historical data or modeling.
234
235
Instead, the risk-based analysis for production or quality system software considers those factors
236
that may impact or prevent the software from performing as intended, such as proper system
237
configuration and management, security of the system, data storage, data transfer, or operation
238
error. Thus, a risk-based analysis for production or quality system software should consider
239
which failures are reasonably foreseeable (as opposed to likely) and the risks resulting from each
240
such failure. This guidance discusses both process risks and medical device risks. A process risk
241
refers to the potential to compromise production or the quality system. A medical device risk
242
refers to the potential for a device to harm the patient or user. When discussing medical device
243

Contains Nonbinding Recommendations
Draft – Not for Implementation
10
risks, this guidance focuses on the medical device risk resulting from a quality problem that
244
compromises safety.
245
246
Specifically, FDA considers a software feature, function, or operation to pose a high process risk
247
when its failure to perform as intended may result in a quality problem that foreseeably
248
compromises safety, meaning an increased medical device risk. This process risk
249
identification step focuses only on the process, as opposed to the medical device risk posed to the
250
patient or user. Examples of software features, functions, or operations that are generally high
251
process risk are those that:
252
253
· maintain process parameters (e.g., temperature, pressure, or humidity) that affect the
254
physical properties of product or manufacturing processes that are identified as essential
255
to device safety or quality;
256
257
· measure, inspect, analyze and/or determine acceptability of product or process with
258
limited or no additional human awareness or review;
259
260
· perform process corrections or adjustments of process parameters based on data
261
monitoring or automated feedback from other process steps without additional human
262
awareness or review;
263
264
· produce directions for use or other labeling provided to patients and users that are
265
necessary for safe operation of the medical device; and/or
266
267
· automate surveillance, trending, or tracking of data that the manufacturer identifies as
268
essential to device safety and quality.
269
270
In contrast, FDA considers a software feature, function, or operation not to pose a high process
271
risk when its failure to perform as intended would not result in a quality problem that
272
foreseeably compromises safety. This includes situations where failure to perform as
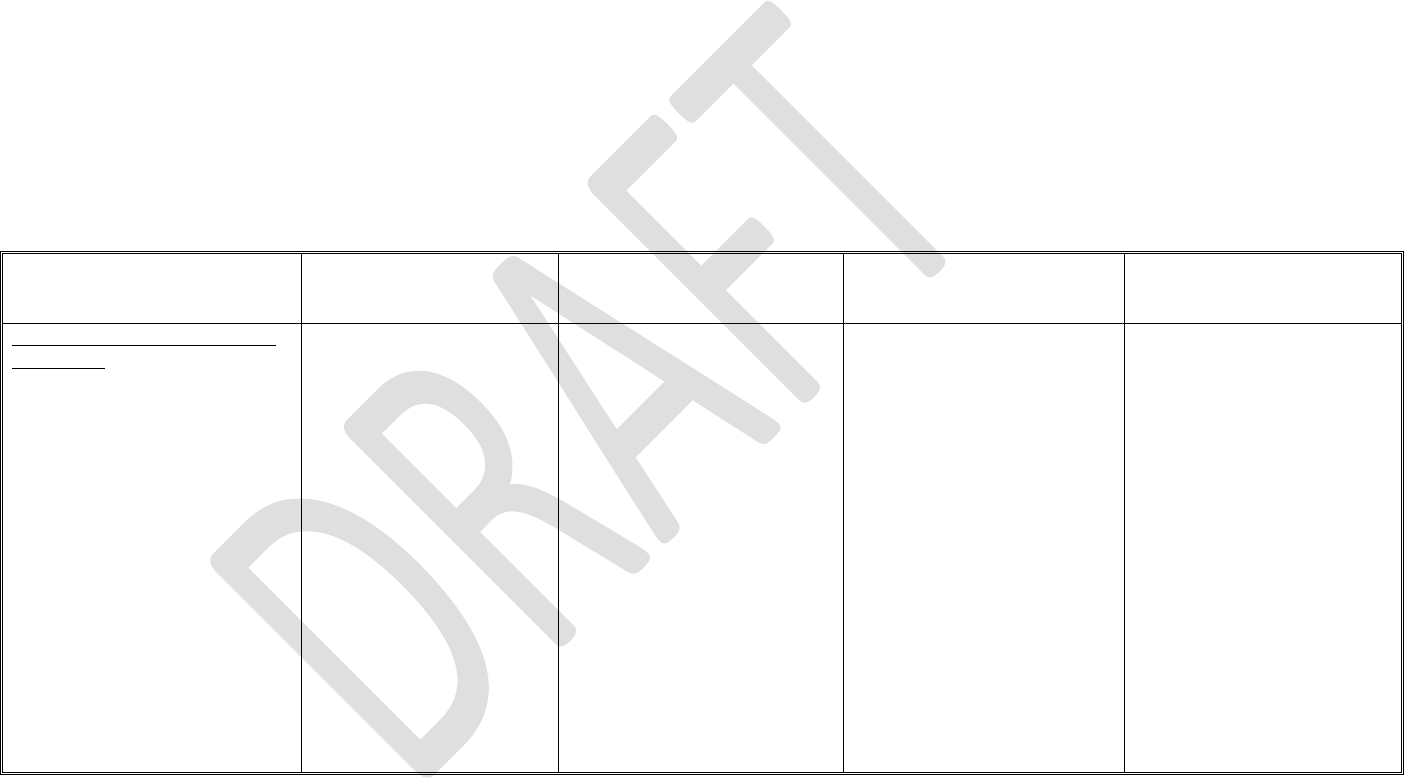
273
intended would not result in a quality problem, as well as situations where failure to
274
perform as intended may result in a quality problem that does not foreseeably lead to
275
compromised safety. Examples of software features, functions, or operations that generally are
276
not high process risk include those that:
277
278
· collect and record data from the process for monitoring and review purposes that do not
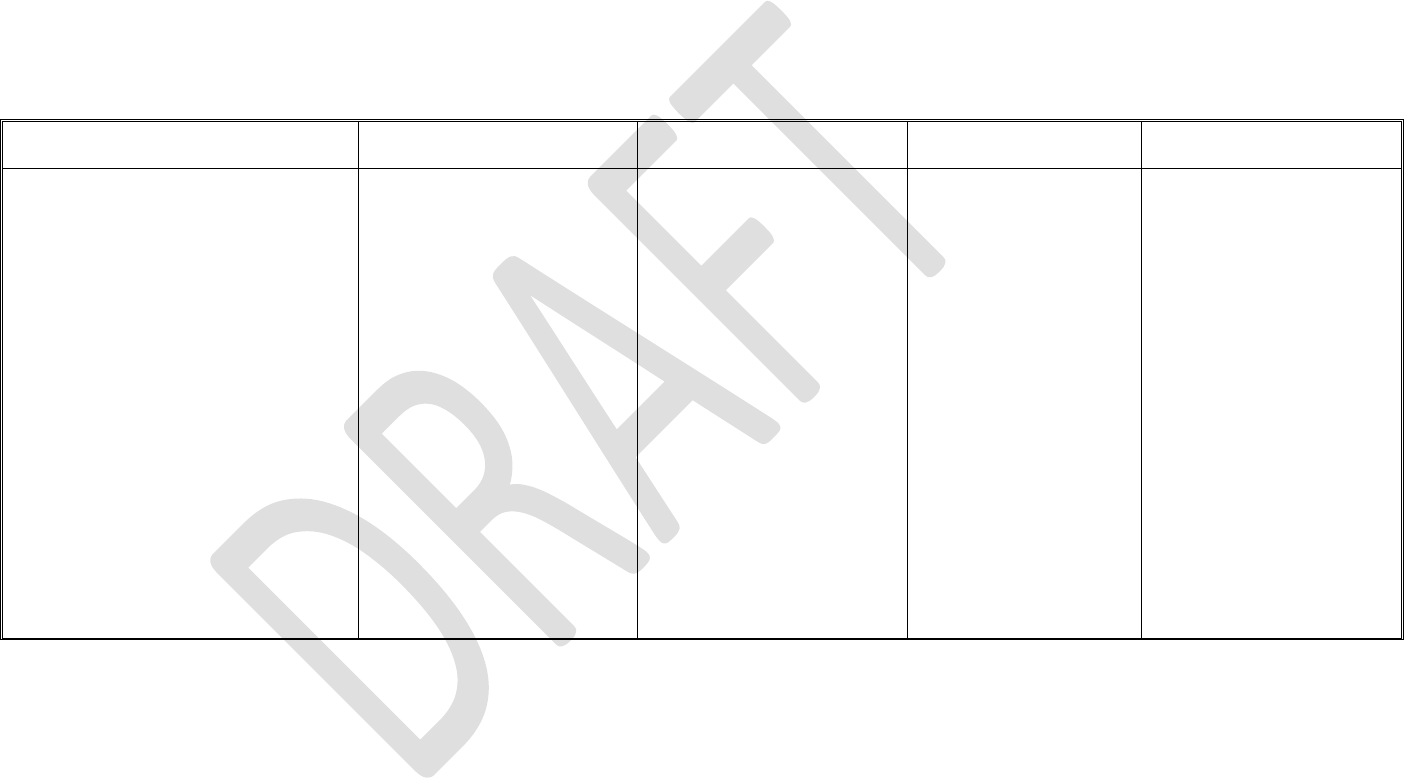
279
have a direct impact on production or process performance;
280
281
· are used as part the quality system for Corrective and Preventive Actions (CAPA)
282
routing, automated logging/tracking of complaints, automated change control
283
management, or automated procedure management;
284
285
· are intended to manage data (process, store, and/or organize data), automate an existing
286
calculation, increase process monitoring, or provide alerts when an exception occurs in an
287
established process; and/or
288

Contains Nonbinding Recommendations
Draft – Not for Implementation
11
289
· are used to support production or the quality system, as explained in Section V.A. above.
290
291
FDA acknowledges that process risks associated with software used as part of production or the
292
quality system are on a spectrum, ranging from high risk to low risk. Manufacturers should
293
determine the risk of each software feature, function, or operation as the risk falls on that
294
spectrum, depending on the intended use of the software. However, FDA is primarily concerned
295
with the review and assurance for those software features, functions, and operations that are high
296
process risk because a failure also poses a medical device risk. Therefore, for the purposes of this
297
guidance, FDA is presenting the process risks in a binary manner, “high process risk” and “not
298
high process risk.” A manufacturer may still determine that a process risk is, for example,
299
“moderate,” “intermediate,” or even “low” for purposes of determining assurance activities; in
300
such a case, the portions of this guidance concerning “not high process risk” would apply. As
301
discussed in Section V.C. below, assurance activities should be conducted for software that is
302
“high process risk” and “not high process risk” commensurate with the risk.
303
304
Example 1: An Enterprise Resource Planning (ERP) Management system contains a feature that
305
automates manufacturing material restocking. This feature ensures that the right materials are
306
ordered and delivered to appropriate production operations. However, a qualified person checks
307
the materials before their use in production. The failure of this feature to perform as intended
308
may result in a mix-up in restocking and delivery, which would be a quality problem because the
309
wrong materials would be restocked and delivered. However, the delivery of the wrong materials
310
to the qualified person should result in the rejection of those materials before use in production;
311
as such, the quality problem should not foreseeably lead to compromised safety. The
312
manufacturer identifies this as an intermediate (not high) process risk and determines assurance
313
activities commensurate with the process risk. The manufacturer already undertakes some of
314
those identified assurance activities so implements only the remaining identified assurance
315
activities.
316
317
Example 2: A similar feature in another ERP management system performs the same tasks as in
318
the previous example except that it also automates checking the materials before their use in
319
production. A qualified person does not check the material first. The manufacturer identifies this
320
as a high process risk because the failure of the feature to perform as intended may result in a
321
quality problem that foreseeably compromises safety. As such, the manufacturer will determine
322
assurance activities that are commensurate with the related medical device risk. The
323
manufacturer already undertakes some of those identified assurance activities so implements
324
only the remaining identified assurance activities.
325
326
Example 3: An ERP management system contains a feature to automate product delivery. The
327
medical device risk depends upon, among other factors, the correct product being delivered to
328
the device user. A failure of this feature to perform as intended may result in a delivery mix-up,
329
which would be a quality problem that foreseeably compromises safety; as such, the
330
manufacturer identifies this as a high process risk. Since the failure would compromise safety,
331
the manufacturer will next determine the related increase in device risk and identify the
332
assurance activities that are commensurate with the device risk. In this case, the manufacturer
333

Contains Nonbinding Recommendations
Draft – Not for Implementation
12
has not already implemented any of the identified assurance activities so implements all of the
334
assurance activities identified in the analysis.
335
336
Example 4: An automated graphical user interface (GUI) function in the production software is
337
used for developing test scripts based on user interactions and to automate future testing of
338
modifications to the user interface of a system used in production. A failure of this GUI function
339
to perform as intended may result in implementation disruptions and delay updates to the
340
production system, but in this case, these errors should not foreseeably lead to compromised
341
safety because the GUI function operates in a separate test environment. The manufacturer
342
identifies this as a low (not high) process risk and determines assurance activities that are
343
commensurate with the process risk. The manufacturer already undertakes some of those
344
identified assurance activities so implements only the remaining identified assurance activities.
345
346
As noted in FDA’s guidance, “30-Day Notices, 135 Day Premarket Approval (PMA)
347
Supplements and 75-Day Humanitarian Device Exemption (HDE) Supplements for
348
Manufacturing Method or Process Changes,”
6
for devices subject to a PMA or HDE, changes to
349
the manufacturing procedure or method of manufacturing that do not affect the safety or
350
effectiveness of the device must be submitted in a periodic report (usually referred to as an
351
annual report).
7
In contrast, modifications to manufacturing procedures or methods of
352
manufacture that affect the safety and effectiveness of the device must be submitted in a 30-day
353
notice.
8
Changes to the manufacturing procedure or method of manufacturing may include
354
changes to software used in production or the quality system. For an addition or change to
355
software used in production or the quality system of devices subject to a PMA or HDE, FDA
356
recommends that manufacturers apply the principles outlined above in determining whether the
357
change may affect the safety or effectiveness of the device. In general, if a change may result in a
358
quality problem that foreseeably compromises safety, then it should be submitted in a 30-day
359
notice. If a change would not result in a quality problem that foreseeably compromises safety, an
360
annual report may be appropriate.
361
362
For example, a Manufacturing Execution System (MES) may be used to manage workflow, track
363
progress, record data, and establish alerts or thresholds based on validated parameters, which are
364
part of maintaining the quality system. Failure of such an MES to perform as intended may
365
disrupt operations but not affect the process parameters established to produce a safe and
366
effective device. Changes affecting these MES operations are generally considered annually
367
reportable. In contrast, an MES used to automatically control and adjust established critical
368
production parameters (e.g., temperature, pressure, process time) may be a change to a
369
manufacturing procedure that affects the safety or effectiveness of the device. If so, changes
370
affecting this specific operation would require a 30-day notice.
371
372
6
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/30-day-notices-135-day-
premarket-approval-pma-supplements-and-75-day-humanitarian-device-exemption.
7
21 CFR 814.39(b), 814.126(b)(1), and https://www.fda.gov/regulatory-information/search-fda-guidance-
documents/annual-reports-approved-premarket-approval-applications-pma.
8
21 CFR 814.39(b), 814.126(b)(1). Changes in manufacturing/sterilization site or to design or performance
specifications do not qualify for a 30-day notice.

Contains Nonbinding Recommendations
Draft – Not for Implementation
13
DeterminingtheAppropriateAssuranceActivities
373
Once the manufacturer has determined whether a software feature, function, or operation poses a
374
high process risk (a quality problem that may foreseeably compromise safety), the manufacturer
375
should identify the assurance activities commensurate with the medical device risk or the process
376
risk. In cases where the quality problem may foreseeably compromise safety (high process risk),
377
the level of assurance should be commensurate with the medical device risk. In cases where the
378
quality problem may not foreseeably compromise safety (not high process risk), the level of
379
assurance rigor should be commensurate with the process risk. In either case, heightened risks of
380
software features, functions, or operations generally entail greater rigor, i.e., a greater amount of
381
objective evidence. Conversely, relatively less risk (i.e., not high process risk) of compromised
382
safety and/or quality generally entails less collection of objective evidence for the computer
383
software assurance effort.
384
385
A feature, function, or operation that could lead to severe harm to a patient or user would
386
generally be high device risk. In contrast, a feature, function, or operation that would not
387
foreseeably lead to severe harm would likely not be high device risk. In either case, the risk of
388
the software’s failure to perform as intended is commensurate with the resulting medical device
389
risk.
390
391
If the manufacturer instead determined that the software feature, function, or operation does not
392
pose a high process risk (i.e., it would not lead to a quality problem that foreseeably
393
compromises safety), the manufacturer should consider the risk relative to the process, i.e.,
394
production or the quality system. This is because the failure would not compromise safety, so the
395
failure would not introduce additional medical device risk. For example, a function that collects
396
and records process data for review would pose a lower process risk than a function that
397
determines acceptability of product prior to human review.
398
399
Types of assurance activities commonly performed by manufacturers include, but are not limited
400
to, the following:
401
402
· Unscripted testing – Dynamic testing in which the tester’s actions are not prescribed by
403
written instructions in a test case.
9
It includes:
404
405
· Ad-hoc testing – A concept derived from unscripted practice that focuses primarily
406
on performing testing that does not rely on large amounts of documentation (e.g., test
407
procedures) to execute.
10
408
409
· Error-guessing – A test design technique in which test cases are derived on the basis
410
of the tester’s knowledge of past failures or general knowledge of failure modes.
11
411
412
9
IEC/IEEE/ISO 29119-1 First edition 2013-09-01: Software and systems engineering – Software testing - Part 1:
Concepts and definitions, Section 4.94.
10
Ibid., Section 5.6.5.
11
Ibid., Section 4.14.

Contains Nonbinding Recommendations
Draft – Not for Implementation
14
· Exploratory testing – Experience-based testing in which the tester spontaneously
413
designs and executes tests based on the tester’s existing relevant knowledge, prior
414
exploration of the test item (including results from previous tests), and heuristic
415
“rules of thumb” regarding common software behaviors and types of failure.
416
Exploratory testing looks for hidden properties, including hidden, unanticipated user
417
behaviors, or accidental use situations that could interfere with other software
418
properties being tested and could pose a risk of software failure.
12
419
420
· Scripted testing – Dynamic testing in which the tester’s actions are prescribed by written
421
instructions in a test case. Scripted testing includes both robust and limited scripted
422
testing.
13
423
424
· Robust scripted testing – Scripted testing efforts in which the risk of the computer
425
system or automation includes evidence of repeatability, traceability to requirements,
426
and auditability.
427
428
· Limited scripted testing – A hybrid approach of scripted and unscripted testing that
429
is appropriately scaled according to the risk of the computer system or automation.
430
This approach may apply scripted testing for high-risk features or operations and
431
unscripted testing for low- to medium-risk items as part of the same assurance effort.
432
433
In general, FDA recommends that manufacturers apply principles of risk-based testing in which
434
the management, selection, prioritization, and use of testing activities and resources are
435
consciously based on corresponding types and levels of analyzed risk to determine the
436
appropriate activities.
14
For high-risk software features, functions, and operations, manufacturers
437
may choose to consider more rigor such as the use of scripted testing or limited scripted testing,
438
as appropriate, when determining their assurance activities. In contrast, for software features,
439
functions, and operations that are not high-risk, manufacturers may consider using unscripted
440
testing methods such as ad-hoc testing, error-guessing, exploratory testing, or a combination of
441
methods that is suitable for the risk of the intended use.
442
443
When deciding on the appropriate assurance activities, manufacturers should consider whether
444
there are any additional controls or mechanisms in place throughout the quality system that may
445
decrease the impact of compromised safety and/or quality if failure of the software feature,
446
function or operation were to occur. For example, as part of a comprehensive assurance
447
approach, manufacturers can leverage the following to reduce the effort of additional assurance
448
activities:
449
450
· Activities, people, and established processes that provide control in production. Such
451
activities may include procedures to ensure integrity in the data supporting production or
452
software quality assurance processes performed by other organizational units.
453
454
12
Ibid., Section 4.16.
13
Ibid., Section 4.37.
14
Ibid., Section 4.35.

Contains Nonbinding Recommendations
Draft – Not for Implementation
15
· Established purchasing control processes for selecting and monitoring software
455
developers. For example, the manufacturer could incorporate the practices, validation
456
work, and electronic information already performed by developers of the software as the
457
starting point and determine what additional activities may be needed. For some lower-
458
risk software features, functions, and operations, this may be all the assurance that is
459
needed by the manufacturer.
460
461
· Additional process controls that have been incorporated throughout production. For
462
example, if a process is fully understood, all critical process parameters are monitored,
463
and/or all outputs of a process undergo verification testing, these controls can serve as
464
additional mechanisms to detect and correct the occurrence of quality problems that may
465
occur if a software feature, function, or operation were to fail to perform as intended. In
466
this example, the presence of these controls can be leveraged to reduce the effort of
467
assurance activities appropriate for the software.
468
469
· The data and information periodically or continuously collected by the software for the
470
purposes of monitoring or detecting issues and anomalies in the software after
471
implementation of the software. The capability to monitor and detect performance issues
472
or deviations and system errors may reduce the risk associated with a failure of the
473
software to perform as intended and may be considered when deciding on assurance
474
activities.
475
476
· The use of Computer System Validation tools (e.g., bug tracker, automated testing) for
477
the assurance of software used in production or as part of the quality system whenever
478
possible.
479
480
· The use of testing done in iterative cycles and continuously throughout the lifecycle of
481
the software used in production or as part of the quality system.
482
483
For example, supporting software, as referenced in Section V.A., often carries lower risk, such
484
that the assurance effort may generally be reduced accordingly. Because assurance activities
485
used “directly” in production or the quality system often inherently cover the performance of
486
supporting software, assurance that this supporting software performs as intended may be
487
sufficiently established by leveraging vendor validation records, software installation, or
488
software configuration, such that additional assurance activities (e.g., scripted or unscripted
489
testing) may be unnecessary.
490
491
Manufacturers are responsible for determining the appropriate assurance activities for ensuring
492
the software features, functions, or operations maintain a validated state. The assurance activities
493
and considerations noted above are some possible ways of providing assurance and are not
494
intended to be prescriptive or exhaustive. Manufacturers may leverage any of the activities or a
495
combination of activities that are most appropriate for risk associated with the intended use.
496
497

Contains Nonbinding Recommendations
Draft – Not for Implementation
16
EstablishingtheAppropriateRecord
498
When establishing the record, the manufacturer should capture sufficient objective evidence to
499
demonstrate that the software feature, function, or operation was assessed and performs as
500
intended. In general, the record should include the following:
501
502
· the intended use of the software feature, function, or operation;
503
· the determination of risk of the software feature, function, or operation;
504
· documentation of the assurance activities conducted, including:
505
· description of the testing conducted based on the assurance activity;
506
· issues found (e.g., deviations, failures) and the disposition;
507
· conclusion statement declaring acceptability of the results;
508
· the date of testing/assessment and the name of the person who conducted the
509
testing/assessment;
510
· established review and approval when appropriate (e.g., when necessary, a
511
signature and date of an individual with signatory authority)
512
513
Documentation of assurance activities need not include more evidence than necessary to show
514
that the software feature, function, or operation performs as intended for the risk identified. FDA
515
recommends the record retain sufficient details of the assurance activity to serve as a baseline for
516
improvements or as a reference point if issues occur.
15
517
518
Table 1 provides some examples of ways to implement and develop the record when using the
519
risk-based testing approaches identified in Section V.C. above. Manufacturers may use
520
alternative approaches and provide different documentation so long as their approach satisfies
521
applicable legal documentation requirements.
522
523
Table 1 – Examples of Assurance Activities and Records
524
Assurance
Activity
Test Plan
Test Results
Record
(Including Digital)
Scripted
Testing:
Robust
· Test objectives
· Test cases
(step-by-step
procedure)
· Expected
results
· Independent
review and
approval of test
cases
· Pass/fail for test
case
· Details regarding
any
failures/deviations
found
· Intended use
· Risk determination
· Detailed report of testing performed
· Pass/fail result for each test case
· Issues found and disposition
· Conclusion statement
· Record of who performed testing and
date
· Established review and approval when
appropriate
15
For the Quality System regulation’s general requirements for records, including record retention period, see 21
CFR 820.180.
Contains Nonbinding Recommendations
Draft – Not for Implementation
17
Assurance
Activity
Test Plan
Test Results
Record
(Including Digital)
Scripted
Testing:
Limited
· Limited test
cases (step-by-
step procedure)
identified
· Expected
results for the
test cases
· Identify
unscripted
testing applied
· Independent
review and
approval of test
plan
· Pass/fail for test
case identified
· Details regarding
any
failures/deviations
found
· Intended use
· Risk determination
· Summary description of testing
performed
· Pass/fail test result for each test case
· Issues found and disposition
· Conclusion statement
· Record of who performed testing and
date
· Established review and approval when
appropriate
Unscripted
Testing:
Ad-hoc
· Testing of
features and
functions with
no test plan
· Details regarding
any
failures/deviations
found
· Intended use
· Risk determination
· Summary description of features and
functions tested and testing performed
· Issues found and disposition
· Conclusion statement
· Record of who performed testing and
date of testing
· Established review and approval when
appropriate
Unscripted
Testing:
Error guessing
· Testing of
failure-modes
with no test
plan
· Details regarding
any failures/
deviations found
· Intended use
· Risk determination
· Summary description of failure-modes
tested and testing performed
· Issues found and disposition
· Conclusion statement
· Record of who performed testing and
date of testing
· Established review and approval when
appropriate
Unscripted
Testing:
Exploratory
Testing
· Establish high
level test plan
objectives (no
step-by-step
procedure is
necessary)
· Pass/fail for each
test plan objective
· Details regarding
any
failures/deviations
found
· Intended use
· Risk determination
· Summary description of the objectives
tested and testing performed
· Pass/fail test result for each objective
· Issues found and disposition
· Conclusion statement
· Record of who performed testing and
date of testing
· Established review and approval when
appropriate
525
526
527
528

Contains Nonbinding Recommendations
Draft – Not for Implementation
18
The following is an example of a record of assurance in a scenario where a manufacturer has
529
developed a spreadsheet with the intended use of collecting and graphing nonconformance data
530
stored in a controlled system for monitoring purposes. In this example, the manufacturer has
531
established additional process controls and inspections that ensure non-conforming product is not
532
released. In this case, failure of the spreadsheet to perform as intended would not result in a
533
quality problem that foreseeably leads to compromised safety, so the spreadsheet would not pose
534
a high process risk. The manufacturer conducted rapid exploratory testing of specific functions
535
used in the spreadsheet to ensure that analyses can be created, read, updated, and/or deleted.
536
During exploratory testing, all calculated fields updated correctly except for one deviation that
537
occurred during update testing. In this scenario, the record would be documented as follows:
538
539
· Intended Use: The spreadsheet is intended for use in collecting and graphing
540
nonconformance data stored in a controlled system for monitoring purposes; as such, it is
541
used as part of production or the quality system. Because of this use, the spreadsheet is
542
different from similar software used for business operations such as for accounting.
543
544
· Risk-Based Analysis: In this case, the software is only used to collect and display data
545
for monitoring nonconformances, and the manufacturer has established additional process
546
controls and inspections to ensure that nonconforming product is not released. Therefore,
547
failure of the spreadsheet to perform as intended should not result in a quality problem
548
that foreseeably leads to compromised safety. As such, the software does not pose a high
549
process risk, and the assurance activities should be commensurate with the process risk.
550
551
· Tested: Spreadsheet X, Version 1.2
552
553
· Test type: Unscripted testing – exploratory testing
554
555
· Goal: Ensure that analyses can be correctly created, read, updated, and deleted
556
557
· Testing objectives and activities:
558
559
o Create new analysis – Passed
560
o Read data from the required source – Passed
561
o Update data in the analysis – Failed due to input error, then passed
562
o Delete data – Passed
563
o Verify through observation that all calculated fields correctly update with changes
564
– Passed with noted deviation
565
566
· Deviation: During update testing, when the user inadvertently input text into an
567
updatable field requiring numeric data, the associated row showed an immediate error.
568
569
· Conclusion: No errors were observed in the spreadsheet functions beyond the deviation.
570
Incorrectly inputting text into the field is immediately visible and does not impact the risk
571
of the intended use. In addition, a validation rule was placed on the field to permit only
572
numeric data inputs.
573

Contains Nonbinding Recommendations
Draft – Not for Implementation
19
574
· When/Who: July 9, 2019, by Jane Smith
575
576
Advances in digital technology may allow for manufacturers to leverage automated traceability,
577
testing, and the electronic capture of work performed to document the results, reducing the need
578
for manual or paper-based documentation. As a least burdensome method, FDA recommends the
579
use of electronic records, such as system logs, audit trails, and other data generated by the
580
software, as opposed to paper documentation and screenshots, in establishing the record
581
associated with the assurance activities.
582
583
Manufacturers have expressed confusion and concern regarding the application of Part 11,
584
Electronic Records; Electronic Signatures, to computers or automated data processing systems
585
used as part of production or the quality system. As described in the “Part 11, Electronic
586
Records; Electronic Signatures – Scope and Application” guidance,
16
the Agency intends to
587
exercise enforcement discretion regarding Part 11 requirements for validation of computerized
588
systems used to create, modify, maintain, or transmit electronic records (see 21 CFR 11.10(a)
589
and 11.30). In general, Part 11 applies to records in electronic form that are created, modified,
590
maintained, archived, retrieved, or transmitted under any records requirements set forth in
591
Agency regulations (see 21 CFR 11.1(b)). Part 11 also applies to electronic records submitted to
592
the Agency under requirements of the Federal Food, Drug, and Cosmetic Act (FD&C Act) and
593
the Public Health Service Act (PHS Act), even if such records are not specifically identified in
594
Agency regulations (see 21 CFR 11.1(b)).
595
596
In the context of computer or automated data processing systems, for computer software used as
597
part of production or the quality system, a document required under Part 820 and maintained in
598
electronic form would generally be an “electronic record” within the meaning of Part 11 (see 21
599
CFR 11.3(b)(6)). For example, if a document requires a signature under Part 820 and is
600
maintained in electronic form, then Part 11 applies (see, e.g., 21 CFR 820.40 (requiring
601
signatures for control of required documents)).
602
16
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/part-11-electronic-records-
electronic-signatures-scope-and-application.
Contains Nonbinding Recommendations
Draft – Not for Implementation
20
AppendixA.Examples603
The examples in this section outline possible application of the principles in this draft guidance to various software assurance 604
situations cases.605
Example1:NonconformanceManagementSystem606
A manufacturer has purchased COTS software for automating their nonconformance process and is applying a risk-based approach for 607
computer software assurance in its implementation. The software is intended to manage the nonconformance process electronically. 608
The following features, functions, or operations were considered by the manufacturer in developing a risk-based assurance strategy:609
610
Table 2. Computer Software Assurance Example for a Nonconformance Management System611
Features, Functions, or
Operations
Intended Use of the
Features, Functions or
Operations
Risk-Based Analysis
Assurance Activities
Establishing the appropriate
record
Nonconformance (NC) Initiation
Operations:
· A nonconforming event
results in the creation of an
NC record.
· The necessary data for
initiation are recorded prior to
completion of an NC
initiation task.
· An NC Owner is assigned
prior to completion of the NC
initiation task.
The intended uses of the
operations are to manage the
workflow of the
nonconformance and to
error-proof the workflow to
facilitate the work and a
complete quality record.
These operations are
intended to supplement
processes established by the
manufacturer for
containment of non-
conforming product.
Failure of the NC initiation
operation to perform as intended
may delay the initiation
workflow, but would not result
in a quality problem that
foreseeably compromises safety,
as the manufacturer has
additional processes in place for
containment of non-conforming
product. As such, the
manufacturer determined the NC
initiation operations did not pose
a high process risk.
The manufacturer has
performed an assessment of the
system capability, supplier
evaluation, and installation
activities. In addition, the
manufacturer supplements these
activities with exploratory
testing of the operations. High
level objectives for testing are
established to meet the intended
use and no unanticipated
failures occur.
The manufacturer documents:
· the intended use
· risk determination,
· summary description of the
features, functions,
operations tested
· the testing objectives and
if they passed or failed
· any issues found and their
disposition
· a concluding statement
noting that the
performance of the
operation is acceptable
· the date testing was
performed, and who
performed the testing.

Contains Nonbinding Recommendations
Draft – Not for Implementation
21
Features, Functions, or
Operations
Intended Use of the
Features, Functions or
Operations
Risk-Based Analysis
Assurance Activities
Establishing the appropriate
record
Electronic Signature Function:
· The electronic signature
execution record is stored as
part of the audit trail.
· The electronic signature
employs two distinct
identification components of
a login and password.
· When an electronic signature
is executed, the following
information is part of the
execution record:
o The name of the person
who signs the record
o The date (DD-MM-
YYYY) and time
(hh:mm) the signature
was executed.
o The meaning associated
with the signature (such
as review, approval,
responsibility, or
authorship).
The intended use of the
electronic signature function
is to capture and store an
electronic signature where a
signature is required and
such that it meets
requirements for electronic
signatures.
If the electronic signature
function were to fail to perform
as intended, then production or
quality system records may not
reflect appropriate approval or
be sufficiently auditable, or may
fail to meet other regulatory
requirements. However, such a
failure would not foreseeably
lead to compromised safety. As
such, the manufacturer
determined that this function
does not pose high process risk.
The manufacturer has
performed an assessment of the
system capability, supplier
evaluation, and installation
activities. To provide assurance
that the function complies with
applicable requirements, the
manufacturer performs ad-hoc
testing of this function with
users to demonstrate the
function meets the intended use.
The manufacturer documents:
· the intended use
· risk determination
· testing performed
· any issues found and their
disposition
· a concluding statement
noting that the
performance of the
function is acceptable
· the date testing was
performed and who
performed the testing.

Contains Nonbinding Recommendations
Draft – Not for Implementation
22
Features, Functions, or
Operations
Intended Use of the
Features, Functions or
Operations
Risk-Based Analysis
Assurance Activities
Establishing the appropriate
record
Product Containment Function:
· When a nonconformance is
initiated for product outside
of the manufacturer’s control,
then the system prompts the
user to identify if a product
correction or removal is
needed.
This function is intended to
trigger the necessary
evaluation and decision-
making on whether a product
correction or removal is
needed when the
nonconformance occurred in
product that has been
distributed.
Failure of the function to
perform as intended would
result in a necessary correction
or removal not being initiated,
resulting in a quality problem
that foreseeably compromises
safety. The manufacturer
therefore determined that this
function poses high process risk.
The manufacturer has
performed an assessment of the
system capability, supplier
evaluation, and installation
activities. Since the
manufacturer determined the
function to pose high process
risk, the manufacturer
determined assurance activities
commensurate with the medical
device risk: established a
detailed scripted test protocol
that exercises the possible
interactions and potential ways
the function could fail. The
testing also included
appropriate repeatability testing
in various scenarios to provide
assurance that the function
works reliably.
The manufacturer documents:
· the intended use
· risk determination
· detailed test protocol
developed
· detailed report of the
testing performed
· pass/fail results for each
test case
· any issues found and their
disposition
· a concluding statement
noting that the
performance of the
operation is acceptable
· the date testing was
performed and who
performed the testing
· the signature and date of
the appropriate signatory
authority.
612
613
614
615
616
Contains Nonbinding Recommendations
Draft – Not for Implementation
23
Example2:LearningManagementSystem(LMS)617
A manufacturer is implementing a COTS LMS and is applying a risk-based approach for computer software assurance in its 618
implementation. The software is intended to manage, record, track, and report on training. The following features, functions, or 619
operations were considered by the manufacturer in developing a risk-based assurance strategy: 620
621
Table 3. Computer Software Assurance Example for an LMS622
Features, Functions, or Operations
Intended Use of the Features,
Functions or Operations
Risk-Based Analysis
Assurance Activities
Establishing the
appropriate record
· The system provides user log-on
features (e.g., username and
password)
· The system assigns trainings to users
per the curriculum assigned by
management
· The system captures evidence of
users’ training completion
· The system notifies users of training
curriculum assignments, completion
of trainings, and outstanding
trainings
· The system notifies users’
management of outstanding trainings
· The system generates reports on
training curriculum assignments,
completion of training, and
outstanding trainings
All of the features, functions,
and operations have the same
intended use, that is, to manage,
record, track and report on
training. They are intended to
automate processes to comply
with 21 CFR 820.25
(Personnel), and to establish the
necessary records.
Failure of these features,
functions, or operations to
perform as intended would
impact the integrity of the
quality system record but
would not foreseeably
compromise safety. As such,
the manufacturer determined
that the features, functions,
and operations do not pose
high process risk.
The manufacturer has
performed an assessment
of the system capability,
supplier evaluation, and
installation activities. In
addition, the manufacturer
supplements these
activities with unscripted
testing, applying error-
guessing to attempt to
circumvent process flow
and “break” the system
(e.g. try to delete the audit
trail).
The manufacturer
documents:
· the intended use
· risk determination
· a summary description
of the failure modes
tested
· any issues found and
their disposition
· a concluding statement
noting that the
performance of the
operation is acceptable
· the date testing was
performed, and who
performed the testing.
623
624
Contains Nonbinding Recommendations
Draft – Not for Implementation
24
Example3:BusinessIntelligenceApplications625
A medical device manufacturer has decided to implement a commercial business intelligence solution for data mining, trending, and 626
reporting. The software is intended to better understand product and process performance over time, in order to provide identification 627
of improvement opportunities. The following features, functions, or operations were considered by the manufacturer in developing a 628
risk-based assurance strategy: 629
630
Table 4. Computer Software Assurance Example for a Business Intelligence Application631
Features, Functions, or
Operations
Intended Use of the
Features, Functions or
Operations
Risk-Based Analysis
Assurance Activities
Establishing the appropriate
record
Connectivity Functions:
· The software allows for
connecting to various
databases in the
organization and external
data sources.
· The software maintains
the integrity of the data
from the original sources
and is able to determine if
there is an issue with the
integrity of the data,
corruption, or problems
in data transfer.
These functions are
intended to ensure a secure
and robust capability for the
system to connect to the
appropriate data sources,
ensure integrity of the data,
prevent data corruption,
modify, and store the data
appropriately.
Failure of these functions to
perform as intended would result
in inaccurate or inconsistent
trending or analysis. This would
result in failure to identify
potential quality trends, issues or
opportunities for improvement,
which in some cases, may result in
a quality problem that foreseeably
compromises safety. As such, the
manufacturer determined that these
functions posed high process risk,
necessitating more-rigorous
assurance activities, commensurate
with the related medical device
risk.
The manufacturer determined
assurance activities
commensurate with the medical
device risk and has performed
an assessment of the system
capability, supplier evaluation,
and installation activities.
Additionally, the manufacturer
establishes a detailed scripted
test protocol that exercises the
possible interactions and
potential ways the functions
could fail. The testing also
includes appropriate
repeatability testing in various
scenarios to provide assurance
that the functions work reliably.
The manufacturer documents:
· the intended use
· risk determination
· detailed test protocol
· a detailed report of the testing
performed
· pass/fail results for each test
case
· any issues found and their
disposition
· a concluding statement
noting that the performance
of the operation is acceptable
· the date testing was
performed, and who
performed the testing
· the signature and date of the
appropriate signatory
authority.

Contains Nonbinding Recommendations
Draft – Not for Implementation
25
Features, Functions, or
Operations
Intended Use of the
Features, Functions or
Operations
Risk-Based Analysis
Assurance Activities
Establishing the appropriate
record
Usability Feature:
· The software provides the
user a help menu for the
application.
This feature is intended to
facilitate the interaction of
the user with the system and
provide assistance on use of
all the system features.
The failure of the feature to
perform as intended is unlikely to
result in a quality problem that
would lead to compromised safety.
Therefore, the manufacturer
determined that the feature does
not pose high process risk.
The feature does not necessitate
any additional assurance effort
beyond what the manufacturer
has already performed in
assessing the system capability,
supplier evaluation, and
installation activities.
The manufacturer documents:
· the intended use
· risk determination
· the date of assessment and
who performed the
assessment
· a concluding statement
noting that the performance
is acceptable given the
intended use and risk.
Reporting Functions:
· The software is able to
create and perform
queries and join data
from various sources to
perform data mining.
· The software allows for
various statistical analysis
and data summarization.
· The software is able to
create graphs from the
data.
· The software provides the
capability to generate
reports of the analysis.
These functions are
intended to allow the user to
query the data sources, join
data from various sources,
perform analysis, and
generate visuals and
summaries. These functions
are intended for collection
and recording data for
monitoring and review
purposes that do not have a
direct impact on production
or process performance. In
this example, the software is
not intended to inform
quality decisions.
Failure of these functions to
perform as intended may result in a
quality problem (e.g., incomplete
or inadequate reports) but, in this
example, would not foreseeably
lead to compromised safety
because these functions are
intended for collection and
recording data for monitoring and
review purposes that do not have a
direct impact on production or
process performance. Therefore,
the manufacturer determined that
these functions do not pose high
process risk.
The supplier of the reporting
software has validated the
ability of the software to create
and perform queries, join data
from various sources to
perform data mining, perform
statistical analysis and data
summarization, create graphs
and generate reports. Beyond
this, the manufacturer has
assessed the system capability
and performed supplier
evaluation and installation
activities. As such, the
manufacturer determined that
the reporting functions of the
software do not necessitate any
additional assurance effort
beyond these activities.
The manufacturer documents:
· the intended use
· risk determination
· the date of assessment and
who performed the
assessment
· a concluding statement
noting that the performance
is acceptable given the
intended use and risk.
632
